首页 / 更多教程 / LAMMPS学习系列(2)
LAMMPS学习系列(2)
内容导读
互联网集市收集整理的这篇技术教程文章主要介绍了LAMMPS学习系列(2),小编现在分享给大家,供广大互联网技能从业者学习和参考。文章包含1748字,纯文字阅读大概需要3分钟。
内容图文
")
LAMMPS学习系列(2)
初始模拟系统设置
LAMMPS提供的命令有很多,其中经常用于初始模拟系统设置的只有4个,分别为 units, boundary, atom_style, 还有一些比较不常用的(不常用的就是设置为默认值)有dimension,neighbor等
对于units命令,手册的介绍为:



简单来说,units命令是用来定义整个模拟系统中的单位的,举个例子:
units metal
使用这条命令,则计算中一切与时间挂钩的量都是以皮秒为单位;
units real
使用这条命令,则计算中一切与时间挂钩的量都是以飞秒为单位;
units命令使用不当会导致后期数据处理需要进行繁杂的单位换算。所以计算前要思考清楚选取何种单位体系最合适。
对于boundary命令,手册的介绍为:

即boundary命令是用来定义边界条件的,LAMMPS提供了四种边界条件分别为:
p:周期性边界条件
f:非周期性边界条件,采用这种边界条件,当有原子运动到盒子以外的区域时,该原子便会被系统删除。需与
thermo_modify lost ignore 这条命令结合使用。
s:该方向的大小会随着原子的运动而改变(以保证不丢失原子)。
m:该方向的大小会随着原子的运动而改变,但有最小值。
对于atom_style命令,手册的介绍为:
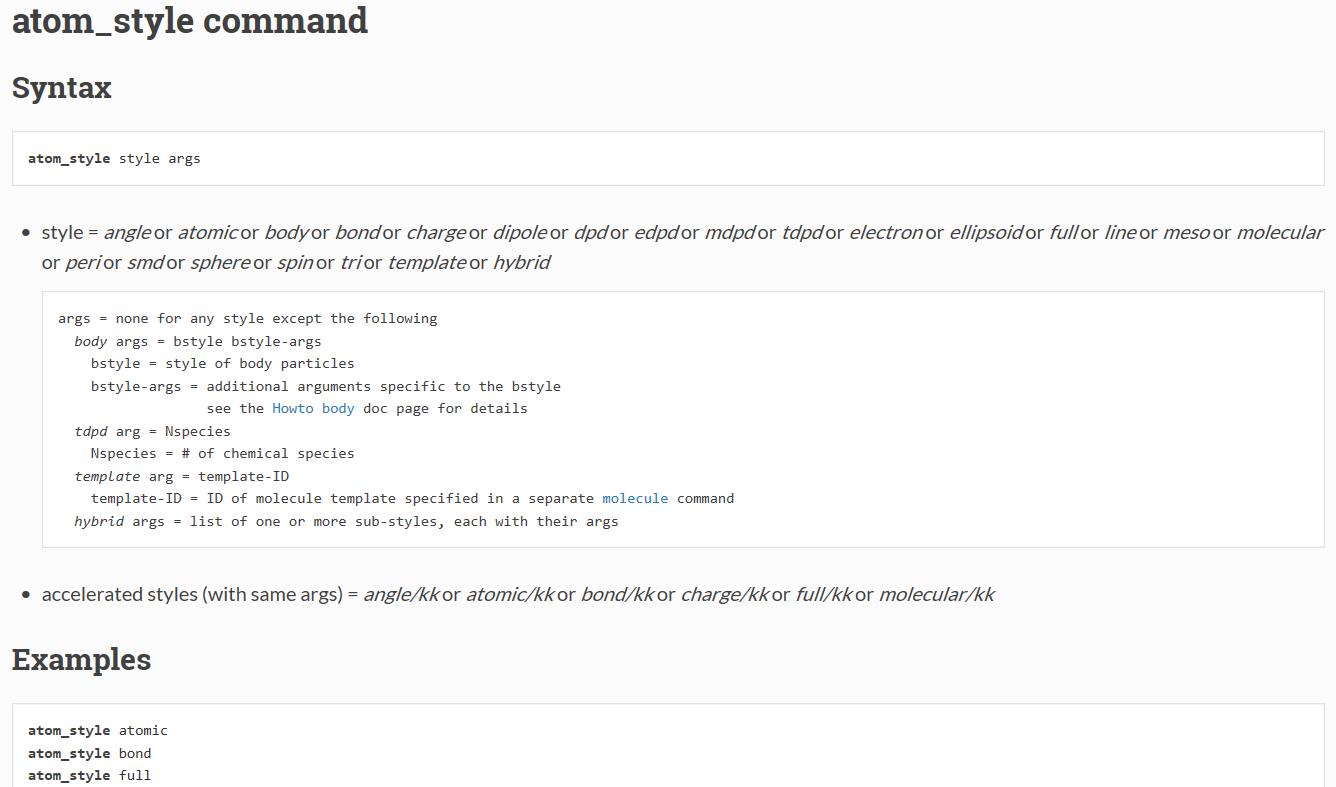
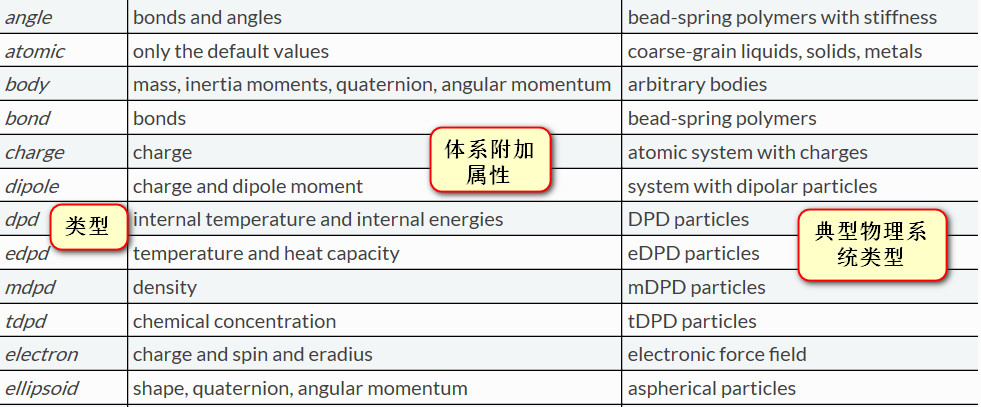
atom_style 这个命令是用来告诉LAMMPS你的模型中有些什么东西比如说原子,键角,电荷之类的,若模型中只有原子:
atom_style atomic (一般金属体系就选这个即可)
若模型中还要考虑键角作用,则:
atom_style angle (可能适合于水分子模型模拟)
LAMMPS在计算各个粒子间作用力时,当两个原子的距离超过截断半径时,它们之间的作用将不被考虑。但是,分子动力学程序在运行时,如果每一步都要判断每个原子的截断半径内有哪些原子,所耗费的计算资源也会随之原子数的增加而指数级增加。为了并行加速计算,LAMMPS构建邻域列表节省原子之间距离的判断的时间。
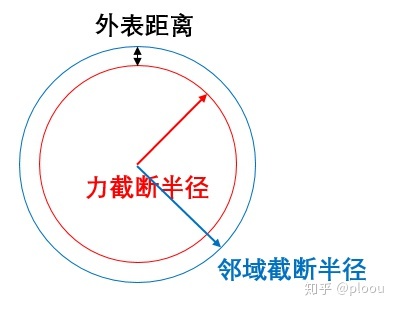
这个命令一般可以不用设置,即保持默认值即可,体系较大时可以适当与neighbor_modify(定义邻域列表的更新频率)修改参数减少计算量(增大bin的值以及降低更新列表的频率。。)
至于dimension命令,这个很简单,定义模拟体系维度的,一般默认为3维(所以一般不用管),也可以设置为2维。
内容总结
以上是互联网集市为您收集整理的LAMMPS学习系列(2)全部内容,希望文章能够帮你解决LAMMPS学习系列(2)所遇到的程序开发问题。 如果觉得互联网集市技术教程内容还不错,欢迎将互联网集市网站推荐给程序员好友。
内容备注
版权声明:本文内容由互联网用户自发贡献,该文观点与技术仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容, 请发送邮件至 gblab@vip.qq.com 举报,一经查实,本站将立刻删除。
内容手机端
扫描二维码推送至手机访问。